• Lysosomes are single-membrane organelles in the cytoplasm. They provide the destruction of complex molecules such as mucopolysaccharide (glycosaminoglycan), sphingolipid and glycoprotein with the enzymes they contain.
• In lysosomal enzyme deficiencies, these complex molecules accumulate and enlarge the lysosomes.
• Organs and tissues involved in lysosomal storage diseases;
- Central nervous system (progressive psychomotor retardation)
- Skeletal system (dysostosis multiplex)
- They involve internal organs (hepatosplenomegaly, pancytopenia).
• Lysosomal storage diseases (LDH) are classified according to the type of complex molecule accumulating in lysosomes (Mucopolysaccharidosis (MPS), Glycosphingolipidosis (GSL), glycoproteinosis (GP))
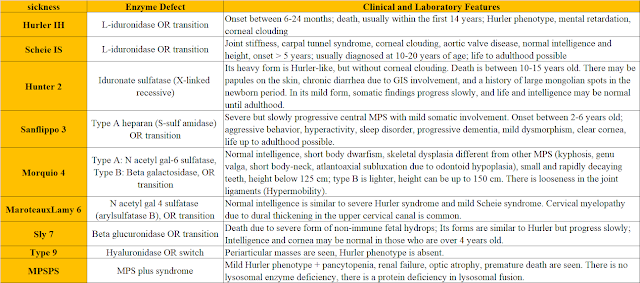
Mucopolysaccharidoses (MPS)
Key features
• Mucopolysaccharides (Glycosaminoglycans), which are structural elements of cartilage, bone, cornea, skin-vascular wall and connective tissues, cannot be destroyed as a result of lysosomal enzyme deficiencies and accumulate in lysosomes.
• Mucopolysaccharidoses show autosomal recessive inheritance, except for MPS-2 (Hunter), which is X-linked inherited.
• Type 3 is the most common, followed by type 1 and 2.
• Metachromatic granulation, called Alder-Reilly Bodies, is observed in leukocytes.
• Skeletal radiographs are requested for diagnosis. Dysostosis multiplex can be shown on the radiographs.
• Urine glycosaminoglycan analysis is done by tandem mass spectrometry.
• Definitive diagnosis is made by demonstrating enzyme deficiency in fibroblasts, leukocytes and serum.
• Enzymotherapy is used in types 1, 2, 4 and 6 in treatment.
• In addition, bone marrow transplantation can be performed in types 1, 2 and 6.
• Enzymotherapy and bone marrow transplantation do not correct neurological findings and mental retardation.
Accumulated MPS and Basic Accumulation Sites
• Heparan sulfate (accumulates in CNS-Psycomotor retardation)
• Dermatan sulfate (accumulates in internal organs-HSM and pancytopenia)
• Keratan sulfate, chondroitin sulfate (accumulate in bone)
Typical Findings
• MMR and growth retardation
• Coarse facial appearance (not seen in type 4) (The most common finding in type 1 MPS patients)
• Macrocephaly, communicating hydrocephalus
• Macroglossia
• Hepatosplenomegaly
• Skeletal deformities (dysostosis multiplex)
• Corneal clouding (not seen in Types 2 and 3)
• Cardiomyopathy and aortic-mitral valve diseases
• Obstructive sleep apnea syndrome, middle ear and sinus infections

Glycoproteinoses

Glycosphingolipidoses (GSL)
Key Features:
• If sphingolipid (SL) is stored only in the periphery and not in the central nervous system, the patient will have hepatosplenomegaly and CI involvement (Gaucher-1).
• If SL deposition is in the central nervous system, mental retardation and neurological findings are prominent. There is no hepatosplenomegaly (Tay-Sachs, Krabbe, MLD).
• If there is involvement of both the central nervous system and peripheral tissue, there is also mental retardation as well as hepatosplenomegaly (Niemann-Pick A, GM1 Gangliosidosis).
• Except for Fabry disease, which is X-linked inherited, it is autosomal recessive.
• Definitive diagnosis is made by showing the missing enzyme in leukocytes and skin fibroblasts and there is no definitive treatment.
• Enzymotherapy is successful in Gaucher-1 and Fabry disease.

GM-1 Gangliosidosis
- OR, 8-Galactosidase deficient, GM1 ganglioside accumulates in neural and visceral cells.
- In addition, keratan sulfate accumulates in the liver
- Mental retardation, hepatosplenomegaly, angiokeratoma
- Dysostosis multiplex similar to mucopolysaccharidoses, coarse face, large tongue, cardiomegaly
- Cherry spot on macula
- Confuses with Hurler syndrome and Niemann Pick-A
Tay-Sachs (GM-2 Gangliosidosis)
- OR, hexosaminidase A deficient
- GM-2 Ganglioside accumulates
- Severe mental retardation (gray matter)
- Cherry spot on macula
- No hepatosplenomegaly
- Initial findings. loss of eye contact, hyperacusis and macrocephaly; convulsions at 2 years old; 4-5 years of death
Niemann-Pick
- OR, Sphingomyelinase deficient
- Type A: Mental retardation (gray matter), spasticity and rigidity, cherry spot in the macula, cirrhosis, hepatosplenomegaly
- Type B: Hepatosplenomegaly, hypersplenism, pulmonary reticulonodular infiltration, mental normal
- Type C: Cholesterol and sphindomyelin transport is impaired, enzyme level is normal/slightly deficient; prolonged history of neonatal jaundice, slowly progressive neurodegenerative disease after 1-2 years of age
- Enzymotherapy in type B and miglustat treatment in type C are continuing.
Wolman's Disease
- Enzymatic acid cholesteryl hydrolase (acid lipase); cholesterol ester and triglyceride accumulation
- Continuous vomiting, steatorrhea, progressive hepatosplenomegaly, hyperlipidemia
- Calcification in the adrenal gland, death around 6 months
Gaucher's Disease
- The most common lipidosis. OR. 8-Glucosidase (glucocerebrosidase) deficient
- Apart from childhood, it can start in adulthood as well.
- Type 1: Glucosyl ceramide accumulates in the reticuloendothelial system. Hepatosplenomegaly, pancytopenia, aseptic necrosis of the femoral head, painful bone lesions, Erlenmeyer deformity in the distal femur, hemorrhages due to thrombocytopenia, storage cells in the bone marrow; there is no central involvement of this type.
- Glucocerebrosidase enzyme replacement is used in treatment. In addition, the glycosyl ceramide synthase inhibitor miglustat is an alternative therapy.
- Type 2: Rapid neurodegenerative findings in addition to visceral involvement, death in the first few years of life
- Type 3: betwen type 1 and type 2; death around age 15
Fabry
- XR, α-Galactosidase deficient; trihexosylceramide (globoside) accumulates.
-Angiokeratomas, painful crises in the extremities, hypohidrosis, corneal opacity
- Ischemic findings in the heart, kidney and brain (ventricular hypertrophy, myocardial infarction, proteinuria and renal failure, cerebral ischemia and infarct) in advancing ages
- Death occurs due to kidney failure or brain/heart involvement.
- Enzymotherapy prevents complications and prolongs life.
Krabbe (Goloboid Cell Leukodystrophy)
- Galkatosylceramide 8-Galactosidase (galactocerebrosidase) deficient.
- White matter is involved in the central nervous system. Attacks of mental retardation, optic atrophy and blindness, spasticity, peripheral neuropathy
- Death in infantile form at age 3
- No hepatosplenomegaly
Farber
- missing enzyme; OR, ceramidase deficient
- Lipogranulomatosis, ceramide accumulates
- Hoarseness, subcutaneous nodules and arthropathy (classic triad) from 3-4 months of age
- Joint deformities similar to rheumatoid arthritis, hepatosplenomegaly
- Neurodegeneration, growth retardation
- Lung accumulation and respiratory failure
Metachromatic Leukodystrophy (MLD)
- OR; Missing enzyme: Aryl sulfatase A
- Difficulty in walking after 1-2 years of age begins with hyperextension of the knee (late infantile form)
- White matter degeneration on MR imaging
- Loss of motor functions, mental retardation, ataxia, speech loss
- Progressive spastic quadriparesis, optic atrophy and death within the first 10 years
- Prolongation of nerve conduction velocity, increase in CSF protein, PAS(+) metachromatic deposits in nerve biopsy and metachromatic granules in urinary sediment are seen.