DEFINITION AND CLASSIFICATION
Biochemical diseases related to protein (enzyme, receptor, transport molecule or membrane element) whose structure or amount is affected as a result of gene mutation are called congenital metabolic diseases. These diseases can be grouped into three main groups:
Intoxication Type Metabolic Disease:
In this type of diseases, symptoms and signs depend on the toxic accumulation of precursors and/or intermediate metabolites due to the block in the intermediate metabolism. Amino acid disorders (phenylketonuria, homocystinuria, tyrosinemia), organic acidemias, urea cycle defects, galactosemia, hereditary fructose intolerance are the main diseases in this group. Initially, infants appear healthy (symptom-free period), as symptoms relate to the degree of accumulation of the toxic substance. While this period is a few days in maple syrup urine disease, symptoms appear towards the end of the first month in phenylketonuria. Symptoms show an acute, acute-recurrent and chronic progressive pattern.
Metabolic Disease Caused by Energy Insufficient:
In this group of diseases, the symptoms depend on the insufficiency of energy production and use in the liver, myocardium, muscle and brain. Glycogen storage diseases, gluconeogenesis defects, congenital lactic acidemias, fatty acid oxidation defects and mitochondrial respiratory disorders are the main diseases in this group. Symptoms occur in acute and acute-intermittent forms (cardiomyopathy, skeletal muscle myopathy, widespread hypotonia, sudden infant death, congenital malformations, hypoglycemia, and hyperlactatemia).
Synthesis and Destruction Disorders of Complex Molecules:
Complex molecules cannot be made or broken down due to enzyme deficiency. Destruction defects are more common. Symptoms are persistent and progressive. Unlike the other 2 groups, they are not affected by intervening factors such as infection and stress (mucopolysaccharidoses, lipid storage diseases, peroxisomal diseases).
Age of onset and mode of appearance of Symptoms
Acute Neonatal Symptoms:
• GI: Inability to weigh in, vomiting, diarrhea
• Cardiopulmonary: Apnea, tachypnea, respiratory distress
• CNS: Lethargy, not sucking, irritability, hypertonia (common), hypotonia (rare), convulsion, coma
• Liver disorders: Jaundice, hepatomegaly, disseminated intravascular coagulopathy (DIC)
• It may give the impression of sepsis in particular. An important feature is that babies look healthy in the first days of life.
Acute-Intermitten Symptoms After the Neonatal Period:
In 1/3 of congenital metabolic diseases, symptoms may occur as recurrent attacks in any period of childhood. Attacks may be accompanied by hypoglycemia, acidosis, ketonuria, lethargy, coma, vomiting, dehydration, ataxia, abdominal pain, psychiatric findings, and myoglobinuria.
Chronic Progressive Symptoms:
Neurological and mental disorders are seen.

Urinary Findings in Congenital Metabolic Diseases (DMD)
Urine Color
- Homogentisic aciduria (alkaptonuria) (black)
- Tryptophan malabsorption (blue)
- Myoglobinuria, porphyria (pink-red)
- Uric acidopathy (yellow-orange)
Urine Smell
- Glutaric acidemia type 2 (sweaty feet)
- Phenylketonuria (mold or dead mouse)
- Maple syrup smelling urine disease (burnt sugar, fenugreek)
- Isovaleric acidemia (sweaty feet)
- Trimethylaminuria (stinky fish)
- Tyrosinemia (rotten cabbage, rancid butter)
- Hyperammonemias (ammonia)
- Hawkinsinuria (swimming pool - chlorine)
- Multiple carboxylase deficiency (male cat urine)
- Hypermethionemia (rotten cabbage)
In general, symptoms and signs that make the diagnosis of congenital metabolic disease suspicious are as follows:
1) Recurrent attacks of vomiting, hypoglycemia, acidosis, odor in urine or sweat, encephalopathy, coma, psychotic behaviors
2) Neurological abnormalities (unexplained mental retardation, developmental arrest, convulsions, dystonia or choreoathetosis)
3) Dysmorphic findings, kidney stones, cardiomyopathy, muscle weakness
4) History of consanguineous marriage and illness-death history in siblings; family history of sudden infant death
ROUTINE LABORATORY TESTS IN CONGENITAL METABOLIC DISEASES (DMH)
Urine Screening Tests
• Abnormal urine odor can best be evaluated by sniffing the tube after keeping the cap closed for 5 minutes at room temperature.
• If three drops of 10% FeCl3 are added to six drops of urine, a dark green color occurs in phenylketonuria, the same test can be used in tyrosinemia, MSUD and histidinemia.
• For the investigation of homocystinuria or cystinuria, a cyclamen pink color is obtained when sodium nitropurisside is dripped onto the urine.
• It is investigated whether there is a reducing substance in the urine with Benedict's solution (carbohydrate metabolism disorders).
• For the diagnosis of MSUD, yellow color occurs in the urine with the 2,4-dinitrophenyl hydrazine (DNPH) test.
• With the addition of NaOH in alkaptonuria, the urine becomes dark brown and black.
Other Tests
• Blood count: There may be neutropenia and thrombocytopenia in organic acidemias.
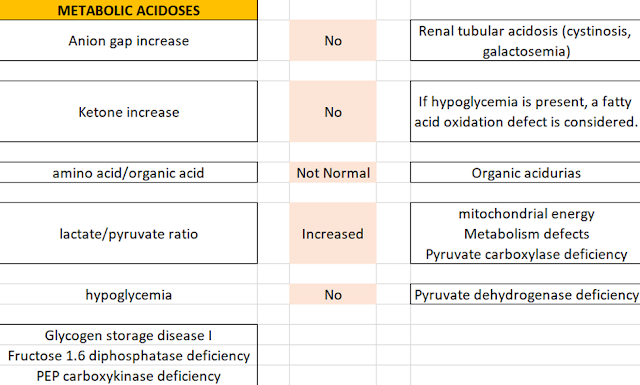
• Blood gases and electrolytes: Required for calculation of metabolic acidosis and anion gap. Anion Gap = [Na+]-[Cl-+HC03 ] (Normal < 12)
• Glucose
• Ammonia: It is necessary for the diagnosis of urea cycle defects and organic acidemia.
• Uric acid: If neurological abnormalities are also present, low uric acid level is diagnostic for molybdenum cofactor deficiency.
• Lactate/Pyruvate: Increased lactate/pyruvate ratio is seen in diseases with energy deficiency and hypoxia. For this reason, it is necessary not to squeeze the arm with a tourniquet while taking blood for lactate.
• Plasma amino acid and acylcarnitine level (with Tandem MS)
• Organic acids in urine: Sampled urine should be stored closed and frozen.
• In general, blood and urine samples should be taken and stored for more specific testing as soon as the patient deteriorates.
Diseases screened in Newborn
Congenital hypothyroidism, phenylketonuria, biotinidase deficiency and cystic fibrosis screenings are included in the routine newborn screening program currently being carried.
General principles in the treatment of metabolic diseases
1. Removal of toxic compounds:
• Mechanical removal (hemodialysis, peritoneal dialysis, exchange transfusion, plasmapheresis)
• Complete removal or reduction of toxic compounds from the diet
• Removal of toxic compounds from the blood with drugs
2. Giving the missing product
3. Increasing enzyme activity (megavitamin therapy)
4. Replacing the missing enzyme (Gaucher, Fabry Disease, MPS I, II, IV and VI, Pompe)
5. Replacing the genetic structure of the diseased cell with a normal cell or correcting the detective gene:
• Organ or tissue transplantation
• Bone marrow transplant (X-linked adrenoleukodystrophy)
• Somatic gene therapy
