GENERAL INFORMATION
• B cell immunodeficiencies can range from a decrease in immunoglobulins to their complete absence.
• Maternal IgG passes through the placenta from the 16th week of gestation. The IgG level at birth is usually slightly higher than that of the mother.
• IgA, IgM, IgD and IgE cannot cross the placenta. These immunoglobulins are too low to be detected in cord blood.
• In the first 4-5 months, IgG decreases, IgM and IgA increase.
• CD19-20-21 are B cell specific antigens.
• B cells constitute 10-20% of peripheral lymphocytes.
Common findings in B cell disorders
• After the 6th month of life, the symptoms start when the effect of passive immunization provided by the antibodies passed from the mother ends.
• Recurrent lower and upper respiratory tract infections are seen with encapsulated bacteria.
• They can usually cope with viral infections. However, paralysis due to live polio vaccine may be fatal CNS infection due to Echovirus.
• Immunoglobulins are low.
• Diarrhea and Giardia infections are common.
• Increased risk of autoimmune disease (Celiac disease) and malignancy.
• Growth retardation is not evident.
• They live for years without complications
Diagnostic tests in B cell disorders
• Immunoglobulin measurement
• Isohemagglutinins: Shows antibodies against A and B group polysaccharide antigens in erythrocytes. These antibodies do not occur in those with AB blood group and under 2 years of age.
• Antibody titers against tetanus, diphtheria, H. influenzae and pneumococcus
• CD19 and CD20 determination
Treatment in B cell disorders
• Bone marrow transplantation is performed in CD-40 ligand defect and X-linked lymphoproliferative syndrome.
• Immunoglobulin is contraindicated in selective IgA deficiency.
• IVIG treatment is applied in other B cell defects
X-linked agammaglobulinemia
• There is a defect in the Bruton tyrosine kinase gene.
• It is common in men. Top 6-9. month is asymptomatic. When the maternally transmitted antibodies are depleted, 6-9. Recurrent diarrhea, most commonly respiratory tract infections (recurrent otitis media, bronchitis, pneumonia), and rarely meningitis, dermatitis, and sometimes arthritis occur in months.
• The most common infectious agents are Streptococcus pneumoniae and Haemophilus influenzae.
• Despite recurrent infections, the diagnosis is approached by hypoplasia of the tonsils, absence of lymphadenopathy and splenomegaly in the child.
Diagnosis
• Diagnosis is made by demonstrating the apparent absence of five immunoglobulins. IgG <100 mg/dl and CD19 B cells <2%. Bowel biopsy or lymph node biopsy is helpful in diagnosis.
• Decreased B cells in peripheral blood is the most important finding in the differential diagnosis of common variable immunodeficiency.
• T cells are normal or increased, their functions are normal. Response to viral infections is normal. However, live viral vaccines should still be avoided. Because the antibody formation response against specific antigens is impaired.
They respond normally to viral infections such as chickenpox and measles. However, in these cases, paralytic poliomyelitis, progressive encephalitis, and fatal echovirus infections may develop after live polio vaccine.
complications
• Meningitis
• Bronchiectasis
• Chronic lung diseases
• Fatal Echovirus and hepatitis infections
• Sinusitis
• Growth hormone deficiency
Treatment
• Gammaglobulins must be administered for life. Gammaglobulin preparations are usually rich in IgG. They contain a small amount of IgM.
Common Variable Immunodeficiency (CVID) Or Acquired Immunodeficiency or Variable Immunodeficiency
• Sporadic, OD or OR can be inherited.
• There is a defect in the transformation of B cells into antibody-making plasma cells and memory B cells.
• Findings occur in advanced ages. Infections are milder than Bruton's.
• The incidence of 1st degree relatives of those with selective IgA deficiency has increased.
• Clinically and laboratoryly, it is mostly confused with Bruton, but it has normal sized tonsils and lymph nodes. 25% of patients have splenomegaly.
• Echoviral meningoencephalitis is very rare.
• Autoimmune diseases are common:
- Celiac-like syndrome may be seen
- Follicular lymphoid hyperplasia in the intestines
- Thymoma
- Alopecia areata
- Pernicious anemia
- Hemolytic anemia
- Gastric atrophy-achlorhydria
- Thrombocytopenia
• Moreover
- Lymphoid interstitial pneumonia
- pseudolymphoma
- B-cell lymphoma
- amyloidosis
- Noncaseating sarcoidosis-like granulomas (lung, spleen, skin and liver) may be seen.
• The risk of malignancy and autoimmune disease increases.
• In those with HIV infection, CVID may improve temporarily or permanently.
• The most common complication is chronic lung disease.
• Anti-IgA antibodies may also be positive and anaphylaxis may be seen in those who have received blood transfusions or those who have received gammaglobulin, just as in selective IgA deficiency.
Diagnosis
• Although B cells are phenotypically normal, there is hypogammaglobulinemia.
• Serum Ig and antibody deficiencies are similar to Bruton's (all Ig levels are decreased). IgA and IgM are either absent or very low.
• Circulating B cell numbers are normal, but upon antigen challenge these B cells cannot differentiate to produce specific antibodies.
• Isohemagglutinins are very low (<1:10)
Treatment
• IVIG, antibiotic
TRANSITIONAL HYPOGAMMAGLOBULINEMIA OF MILK CHILD
• Generally in all infants, 5-6. There is mild hypogammaglobulinemia per month.
• If the child cannot start making his own IgG in the first months, "transient hypogammaglobulinemia" develops and lasts for about a year.
• T and B lymphocyte counts are normal in these patients. T lymphocyte functions are normal. There is a temporary defect in the transformation of B lymphocytes into antibody-secreting plasma cells. The immune response to the vaccine is normal.
• Most of the patients are asymptomatic. However, the frequency of respiratory tract infections may increase in some cases.
• Normally, intravenous immunoglobulin therapy is not performed. However, it can be given in cases with severe infection.
SELECTIVE IGA DEFICIENCY
• It can be inherited autosomal recessively, dominantly and sporadically.
• There may be a deletion in the 18th chromosome.
• It is the most common immunoglobulin deficiency. Cellular immunity is normal.
• The number of B lymphocytes in the circulation is normal. However, there is a defect in the synthesis and secretion of IgA.
• Because anti-IgA antibodies are often positive, anaphylaxis is seen in over-transfused or gammaglobulin recipients.
• Recurrent sinopulmonary (right middle lobe pneumonia), GIS (Giardiasis infection) and urinary system infections, allergic reactions are common.
• Frequency of autoimmune diseases increases with selective IgA deficiency. SLE, rheumatoid arthritis, dermatomyositis, pernicious anemia, Sjögren's syndrome, celiac disease, chronic active hepatitis are the most common autoimmune diseases.
• Increased risk of malignancy. Reticulum cell sarcoma, squamous cell carcinoma of the esophagus and lung, and thymoma are more common in these patients.
• IgG2 deficiency can also be seen in 20% of patients.
• CVID may develop in some of the patients during their follow-up.
• Acquired IgA deficiency or insufficiency can be seen after toxoplasma and measles infections and in those receiving phenytoin or penicillamine therapy.
Diagnosis
• IgA level is less than 10 mg/dl or absent in these patients.
• Peripheral B cell count is normal.
• Cellular immunity is usually normal.
• Antibody response to specific antigens is normal.
• Half of the patients have autoantibodies against IgA.
• It is the most common immunodeficiency associated with allergic diseases.
• They also often contain antibodies to cow's milk protein.
Treatment
• Gammaglobulin is contraindicated.
• Those with IgG2 deficiency have lower levels of anti-IgA antibodies.
• Broad-spectrum antibiotics are used in recurrent sinopulmonary infections.
• Blood products taken from red blood cells or other IgA deficient individuals can be given.
lgG SUBGROUP DEFECTS
• In patients with IgG subgroup deficiency, a clinical picture such as panhypogammaglobulinemia occurs. But lab results show normal Ig levels. IgG subgroup studies confirm the diagnosis.
• There are 4 isotypes of IgG.
- lgGl: 65%
- IgG2: 20%
- IgG3: 10%
- IgG4: 10%
• IgGl and IgG3 against protein antigens
• IgG2 and IgG4 are performed against polysaccharide antigens.
• Even if the serum total IgG level is found to be normal, IgG subgroup deficiency may be found.
• T cell system is normal.
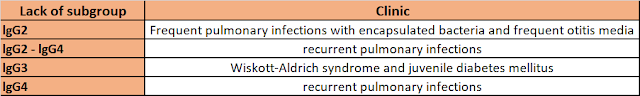
• IgG2 deficiency is most common, followed by IgG4 deficiency.
• Most patients with IgG2 deficiency have IgA deficiency.
• Patients have insufficient antibody responses and severe infections.
• In IgG2 deficiency, frequent infections with polysaccharide capsule bacteria (pneumococci, meningococci and H. influenzae) occur. These patients are unable to produce an antibody response to polysaccharide antigens. All patients with IgG2 deficiency should be vaccinated against H. influenzae Type-B.
Treatment
• Gammaglobulin therapy is used in IgG1 and IgG3 deficiencies.
HIPER-lgM SYNDROME
• The responsible mutations are in 2 genes on the X chromosome (CD40L defect and NEMO defect) and 3 genes on the autosomal chromosomes (Types 2- 3- 4), and all are OR.
• No transition from IgM to A and G (class switch recombination defect).
• Hyper IgM Type 1: It is seen as a result of CD40L mutation and is inherited X-linked. In addition, NEMO mutations cause X-linked Hyper IgM syndrome and are seen together with anhidrotic ectodermal dysplasia.
• Hyper IgM Type 2: It is seen as a result of AICDA (activation-triggered cytidine deaminase) mutation and OR is inherited.
• Hyper IgM Type 3: It is seen as a result of CD40 mutation and OR is inherited. It is very similar to clinical Type-1. The defect is in the B cells.
• Hyper IgM Type 4: The gene is not identified.
• Hyper IgM Type 5: Uracil DNA glycosylase (UNG) defect also causes Hyper IgM syndrome.
Hyper IgM Type1
• As a result of the CD40 ligand (CD154) mutation on activated T cells, T cells cannot send appropriate signals to B cells and B cells only make IgM.
• IgG and A levels are very low, IgM levels are usually normal or sometimes high.
• It becomes symptomatic between the ages of 1-2 after birth.
• Blood B lymphocyte counts are normal. The disorder is in the T cells.
• Tonsillar small, no palpable lymph nodes.
• Recurrent pyogenic infections (otitis media, sinusitis, pneumonia and tonsillitis) are seen.
• The incidence of P. carinii pneumonia and Cryptosporidium enteritis, verruca vulgaris and malignancy has increased.
• There is marked neutropenia.
• Although the circulating B and T cell counts are normal in these patients, the CD27 memory B cell count is decreased (the CD27 B cell count is also decreased in Hyper IgM Type3, while it is normal in other types).
Treatment
• HLA-matched stem cell transplantation, monthly IVIG infusion, and G-CSF for neutropenia.
Hyper lgM Type2
• Unlike Type1, IgM level is very high.
• T cell function tests are normal in contrast to types 1 and 3.
• There is excessive lymphoid hyperplasia.
• They are not susceptible to P. carinii pneumonia and can synthesize isohemagglutinin
• Neutropenia is rarer and usually autoimmune.
• They are prone to develop autoimmune and inflammatory diseases (Diabetes mellitus, polyarthritis, autoimmune hepatitis, hemolytic anemia, immune thrombocytopenia, Crohn's disease and chronic uveitis).
• Monthly IVIG treatment and antibiotic treatment of infections are the main treatment approaches. The clinical course is more benign than Tip 1.
